Rare Disease
Duchenne-Muskeldystrophie
Über Muskeldystrophien
Muskeldystrophien (MD) sind eine Gruppe von mehr als 30 genetisch bedingten Krankheiten, die sich bei den Patienten klinisch als fortschreitende Muskelschwäche mit einem damit verbundenen Verlust an Mobilität, Beweglichkeit und Körperbewegungen äußern. Einige Formen der MD treten bereits im Säuglings- oder Kindesalter auf, während andere erst im mittleren Alter oder später auftreten. Die Duchenne-Muskeldystrophie (DMD) ist die häufigste Form der MD und betrifft hauptsächlich Jungen. Sie wird durch eine Mutation im X-chromosomalen Dystrophin-Gen, das das Protein Dystrophin kodiert, verursacht, einem Protein, das unter anderem an der Aufrechterhaltung der Integrität der Muskeln beteiligt ist.1

1: Mercuri, E. and F. Muntoni (2013). „Muscular dystrophies.“ The Lancet 381(9869): 845-86.
Duchenne-Muskeldystrophie
Die Duchenne-Muskeldystrophie (DMD) ist eine x-chromosomal vererbte, schwere, fortschreitende neuromuskuläre Erbkrankheit, von der weltweit ca. eine von 3.600 männlichen Geburten betroffen ist. DMD tritt auf, wenn eine Mutation im Dystrophin-Gen die Zelle daran hindert, ein funktionsfähiges Dystrophin-Protein zu bilden. Dystrophin spielt eine entscheidende Rolle für die Struktur und die Membranstabilität der Muskelfasern im Skelett-, Zwerchfell- und Herzmuskel. Das Fehlen oder der Mangel an Dystrophin bei Jungen und Männern mit DMD führt zu einer übermäßigen Schädigung der Muskelzellen während der normalen Kontraktions- und Dehnungsaktivität, die schließlich durch Bindegewebe und Fett ersetzt werden, was zu einem fortschreitenden Funktionsverlust führt.
DMD wird in der Regel zwischen dem 2. und 5. Lebensjahr diagnostiziert, wenn Symptome wie Sprachentwicklungsstörungen, motorische Entwicklungsverzögerungen (einschließlich Schwierigkeiten beim Gehen, Treppensteigen und vom Boden aufstehen), oder eine Pseudohypertrophie der Wadenmuskulatur sichtbar werden. Die Muskeldegeneration führt im Alter von ca. 8-14 Jahren unter anderem zum Verlust des Gehvermögens und betrifft nach und nach die oberen Gliedmaßen, die Nackenmuskulatur und andere Bereiche. Der vorzeitige Tod tritt in der Regel im Alter von 20-30 Jahren aufgrund von Komplikationen der Atemwege und des Herzens ein.2
2: Bushby, K., R. Finkel, D. J. Birnkrant, L. E. Case, P. R. Clemens, L. Cripe, A. Kaul, K. Kinnett, C. McDonald, S. Pandya, J. Poysky, F. Shapiro, J. Tomezsko and C. Constantin (2010). „Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management.“ The Lancet Neurology 9(1): 77-93. And „Diagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary care.“ Lancet Neurol 9(2): 177-189.
DMD Krankheitsverlauf (natürlicher Verlauf)¹ ² ³
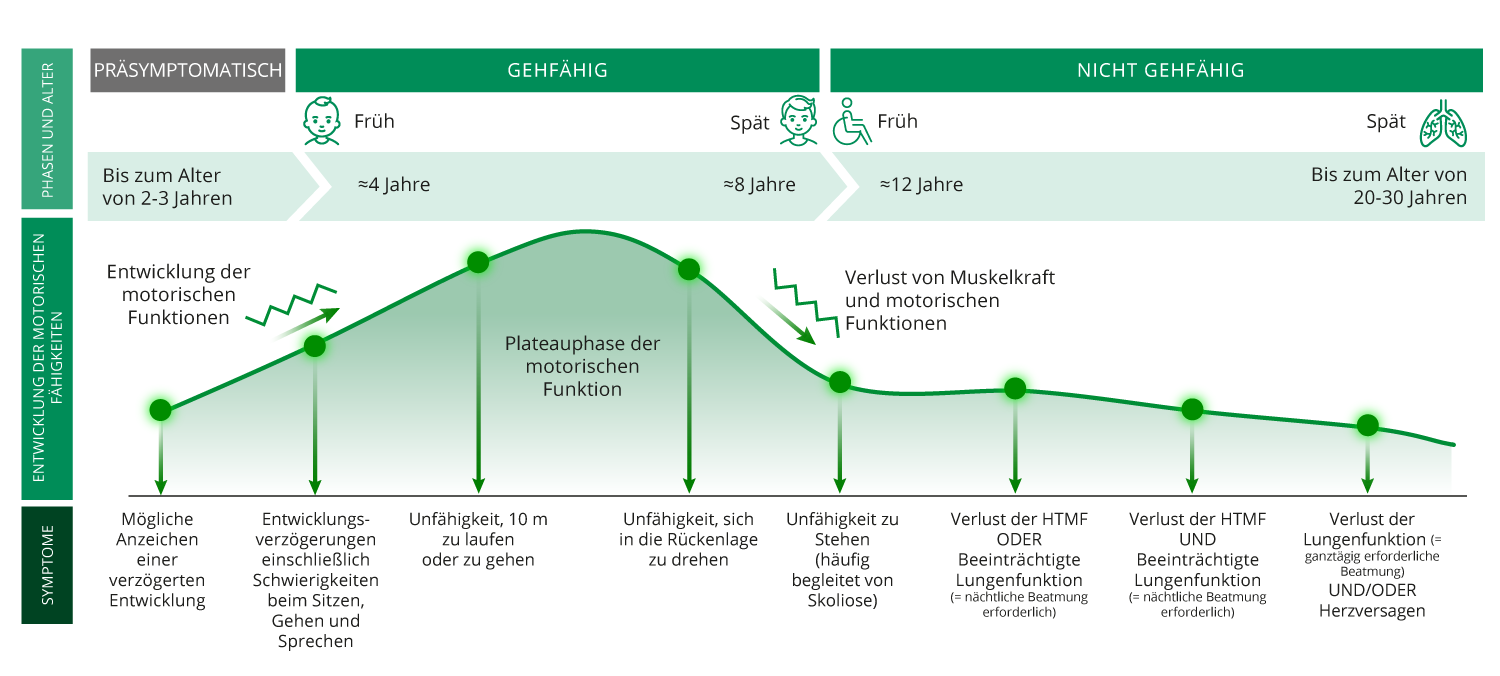
DMD: Duchenne Muskel Dystrophie; HTMF: Hand-to-Mouth-Function.
1. Broomfield J, Hill M, Guglieri M, Crowther M, Abrams K. Life Expectancy in Duchenne Muscular Dystrophy: Reproduced Individual Patient Data Meta-analysis. Neurology. 2021;97(23):e2304-e2314.
2. Landfeldt E, Lindgren P, Bell CF, et al. The burden of Duchenne muscular dystrophy: an international, cross-sectional study. Neurology. 2014;83(6):529-536.
3. Muntoni F, Signorovitch J, Sajeev G, et al. DMD Genotypes and Motor Function in Duchenne Muscular Dystrophy: A Multi-institution Meta-analysis With Implications for Clinical Trials. Neurology. 2023;100(15):e1540-e1554.
Becker-Muskeldystrophie
Die Becker-Muskeldystrophie (BMD) ist eine mildere und variablere Form der Dystrophinopathie mit einer geschätzten Häufigkeit von 1/18.000 bis 1/31.000 männlichen Geburten weltweit. Während bei Mutationen, die die Duchenne Muskeldystrophie verursachen, kein oder sehr geringe Mengen an funktionsfähigem Dystrophin gebildet werden, bilden die Zellen von Menschen mit BMD Dystrophin, das teilweise funktionsfähig ist. Sie stellen eine verkürzte Form des Proteins her, die die Muskeln von BMD-Patienten davor schützt, so vollständig oder so schnell zu degenerieren, als die von Menschen mit DMD. Ähnlich wie bei DMD zeigen Jungen mit BMD eine fortschreitende Muskelschwäche und Pseudohypertrophie der Waden. BMD beginnt in der Regel in der späten Kindheit oder im Jugendalter, zwischen 5 und 15 Jahren, und der Verlauf ist langsamer und weniger vorhersehbar als bei DMD. Die generalisierte Schwäche betrifft zunächst die Muskeln der Hüften, des Beckenbereichs, der Oberschenkel und der Schultern. In einigen Fällen ist eine Herzbeteiligung (Kardiomyopathie) das erste Anzeichen.
Bei der Becker-Muskeldystrophie kommt es in der Regel zu einem langsamen Abbau der Muskelfasern, das Fortschreiten der Krankheit kann bei den Betroffenen unterschiedlich verlaufen. Einige Patienten sind auf einen Rollstuhl angewiesen, während andere vielleicht nur Gehhilfen wie z.B. Stöcke benötigen. Die Lebenserwartung ist aufgrund von Herzerkrankungen und Atemwegskomplikationen oft verkürzt. Die meisten Menschen mit BMD überleben bis ins mittlere oder späte Erwachsenenalter.3
3: Darras, B. T., Miller. D. T. & Urion, D. K. Dystrophinopathies in GeneReviews® (ed Pagon, R. A. et al.). Internet. Seattle (WA): University of Washington, 1993–2015. Available from www.ncbi.nlm.nih.gov/books/NBK1119/
Klinische Arzneimittelstudien
Klinische Studien (syn. klinische Prüfung) sind Forschungsstudien an menschlichen Patienten zum Nachweis der Sicherheit und Wirksamkeit einer experimentellen Therapie oder Behandlung. Bevor klinische Prüfungen von Arzneimitteln an Menschen durchgeführt werden, werden mehrere präklinische Tests im Labor und/oder an Tieren durchgeführt, um zu ermitteln:
- ob das Arzneimittel sicher und wirksam ist
- welches die beste Verabreichungsform ist
- wie es im Körper wirkt
- wie die Nebenwirkungen aussehen

Eine klinische Prüfung ist eine Forschungsstudie, die es ermöglicht, die Informationen zu sammeln, die erforderlich sind, um festzustellen, ob ein Prüfpräparat als neue potenzielle Behandlungsmethode bei Menschen eingesetzt werden kann, und um die Risiken und Vorteile des Prüfpräparats zu bewerten. In der Regel sind drei Phasen erforderlich, um eine neue Behandlung zu entwickeln. Jede Studienphase hat einen anderen Zweck und dient der Beantwortung unterschiedlicher Fragen:
- Phase 1: Ein Studienmedikament wird an einer kleinen Gruppe von Personen, häufig an Freiwilligen ohne Krankheit, getestet, um seine Sicherheit zu bewerten und eine sichere Dosierung zu bestimmen, die verabreicht werden kann.
- Phase 2: Das Prüfpräparat wird an einer größeren Gruppe von Personen getestet, um seine Sicherheit zu bestätigen sowie die Wirksamkeit und mögliche Nebenwirkungen zu bewerten.
- Phase 3: Das Prüfpräparat wird in einer größeren Gruppe und über einen längeren Zeitraum getestet, um seine Wirksamkeit zu bestätigen und weniger häufige oder seltene Nebenwirkungen zu erkennen.
Weitere Informationen über klinische Studien im Allgemeinen finden Sie unter ClinicalTrials.gov ( www.clinicaltrials.gov ) oder Clinical Trial Register EU (https://www.clinicaltrialsregister.eu/ctr-search/search) .
Wenn Sie an der Teilnahme an einer klinischen Studie von Italfarmaco interessiert sind und jemanden kontaktieren möchten, senden Sie bitte eine E-Mail an patientadvocacy@italfarmacogroup.com . Wenn Sie bereits an einer klinischen Prüfung mit Italfarmaco teilnehmen und ein Problem mit dem Prüfpräparat oder eine Nebenwirkung melden möchten, wenden Sie sich bitte so bald wie möglich an Ihren Studienarzt.
Italfarmacos Position zum „Compassionate Use Program“ (Härtefallprogramm)
Die Überzeugung von Italfarmaco ist, dass jeder Mensch Zugang zu einer Gesundheitsversorgung verdient, die es ihm ermöglicht, sein Leben in vollen Zügen zu genießen. Wir haben uns verpflichtet, Menschen, die von schweren und lebensbedrohlichen Krankheiten betroffen sind, durch die Entdeckung, Entwicklung und Inverkehrbringung von Medikamenten zu helfen. Unser Ziel ist es, so vielen Patienten wie möglich, sichere und wirksame Medikamente zur Verfügung zu stellen, indem wir klinische Studien durchführen und die Marktzulassung durch die Zulassungsbehörden ermöglichen. Daher geben wir dem Zugang zu unseren Präparaten in der klinischen Entwicklung durch die Teilnahme an einer klinischen Studie den Vorrang. Eine Liste der klinischen Studien, für die Italfarmaco derzeit Patienten rekrutiert, finden Sie unter www.clinicaltrials.gov.
Wir sind uns bewusst, dass es Umstände gibt, unter denen Patienten mit schweren oder lebensbedrohlichen Krankheiten nicht an einer klinischen Studie teilnehmen können oder dürfen und keinen Zugang zu einem derzeit zugelassenen Medikament haben oder nicht zufriedenstellend damit behandelt werden können. In solchen Situationen ist es manchmal im Interesse der Patienten, Zugang zu Arzneimitteln zu haben, bevor diese zugelassen sind. Italfarmaco hat daher die Teilnahme am nationalen „Härtefallprogramm“ („Compassionate Use Programm“) geprüft.
Wie erfahren Sie mehr über das Härtefallprogramm?
Weitere Informationen und eine Liste mit Medikamenten, die im Rahmen eines Härtefallprogrammes genehmigt wurden, finden Sie auf der Webseite des Bundesinstituts für Arzneimittel und Medizinprodukte.
Unser derzeitiger Ansatz für den frühzeitigen Zugang, wie oben beschrieben, steht im Einklang mit den EURODIS-Empfehlungen an die Industrie für den frühzeitigen Zugang zu Arzneimitteln in Europa, den Empfehlungen der Europäischen Arzneimittelagentur zum „compassionate use“ und den nationalen Richtlinien.